Mechanisms of Proteinuria in HIV
 Gentzon Hall1,2
Gentzon Hall1,2  Christina M. Wyatt1,3*
Christina M. Wyatt1,3*- 1Department of Medicine, Division of Nephrology, Duke University School of Medicine, Durham, NC, United States
- 2Duke Molecular Physiology Institute, Durham, NC, United States
- 3Duke Clinical Research Institute, Durham, NC, United States
Proteinuria is common in the setting of HIV infection, and may reflect comorbid kidney disease, treatment-related nephrotoxicity, and HIV-related glomerular diseases. The mechanisms of podocyte and tubulointerstial injury in HIV-associated nephropathy (HIVAN) have been the subject of intense investigation over the past four decades. The pathologic contributions of viral gene expression, dysregulated innate immune signaling, and ancestry-driven genetic risk modifiers have been explored in sophisticated cellular and whole animal models of disease. These studies provide evidence that injury-induced podocyte dedifferentiation, hyperplasia, cytoskeletal dysregulation, and apoptosis may cause the loss of glomerular filtration barrier integrity and slit diaphragm performance that facilitates proteinuria and tuft collapse in HIVAN. Although the incidence of HIVAN has declined with the introduction of antiretroviral therapy, the collapsing FSGS lesion has been observed in the context of other viral infections and chronic autoimmune disorders, and with the use of interferon-based therapies in genetically susceptible populations. This highlights the fact that the lesion is not specific to HIVAN and that the role of the immune system in aggravating podocyte injury warrants further exploration. This review will summarize our progress in characterizing the molecular mechanisms of podocyte dysfunction in HIVAN and other forms of HIV-associated kidney disease.
Introduction
In the four decades since the first cases of AIDS were reported in 1981, an estimated 77.5 million people have been infected with HIV and more than 34 million people have died from complications of HIV infection (1). Kidney disease emerged as an important complication of HIV in the early years of the epidemic, with the first reports of a unique pattern of collapsing focal segmental glomerulosclerosis (FSGS) with accompanying tubulointerstitial injury published in 1984 (2, 3). HIIV-associated nephropathy (HIVAN) quickly became the leading cause of end-stage kidney disease (ESKD) in people living with HIV (PLWH), demonstrating a marked predilection for individuals of African descent. Although the incidence of ESKD attributed to HIVAN plateaued in the United States following the widespread introduction of 3-drug antiretroviral therapy (ART) in 1997, HIVAN remains an important cause of kidney disease in the setting of untreated HIV infection (4). The original case series also reported a spectrum of immune complex glomerular lesions, and contemporary biopsy series continue to identify immune complex kidney diseases as one of the most common histologic diagnoses in PLWH. Other common causes of kidney disease in PLWH include ART toxicity and comorbid kidney disease due to traditional risk factors such as diabetes (4). As a result, kidney biopsy is often required for definitive diagnosis of proteinuric kidney disease in PLWH.
The epidemiology of HIVAN suggested that both viral and host factors play a central role in pathogenesis. The development of HIVAN in HIV-transgenic mouse models has allowed for extensive investigation into the mechanisms of glomerular injury, proteinuria, and kidney failure in HIVAN, which will be the primary focus of this review.
HIV Infection of the Kidney
The emergence of HIVAN in the setting of AIDS and the decline in incidence of ESKD with the introduction of ART is consistent with a direct role for HIV in the pathogenesis of HIVAN. Early reports demonstrated the presence of HIV nucleic acids in renal epithelial cells (5–7); however, the absence of CD4, CXCR4, and CCR5 receptor expression on these cells (8, 9) implied the existence of a non-receptor mediated viral entry mechanism (10, 11). The subsequent identification of HIV-1 entry into human podocytes via lipid rafts (12) and by dynamin-mediated endocytosis (13–15) provided compelling evidence for non-canonical routes of viral particle entry. Although many questions remain, substantial progress has been made in characterizing the effects of HIV infection on podocyte physiology and function (Figures 1, 2).
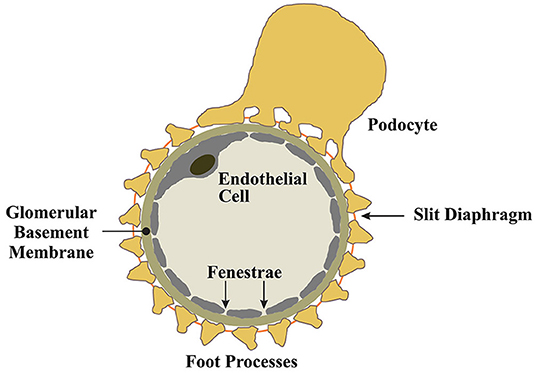
Figure 1. Mechanisms of proteinuria in HIVAN.
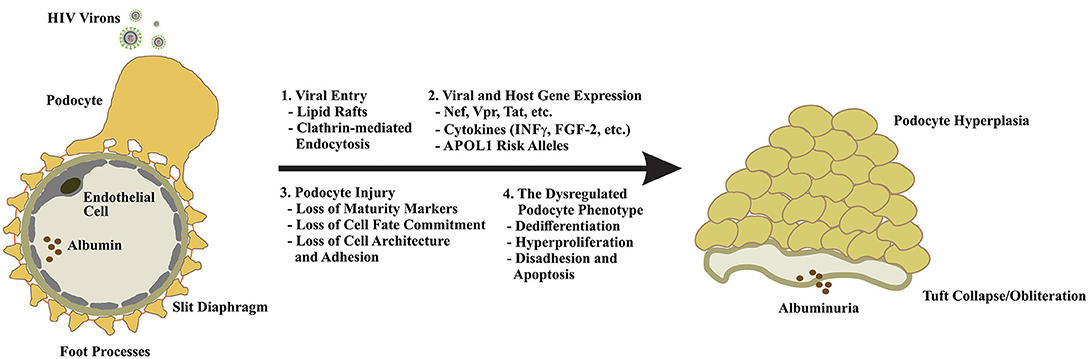
Figure 2. Podocyte dysfunction in response to HIV infection is multifaceted. Because podocytes lack the receptors for canonical viral entry, viral particle entry via lipid rafts, and by clathrin-mediated endocytosis have been proposed (1). Upon entry, expression of viral genes and other mediators of innate immunity drive podocyte cytotoxicity and may enhance the expression of the high-risk APOL1 alleles in individuals of African descent to increase their risk of developing HIVAN (2). Podocyte injury manifests in the loss of maturity markers (e.g., nephrin, synaptopodin, WT1, CALLA, etc.), loss of cell fate commitment, and hyperplasia and the loss of cytoarchitecture and adhesion (3). The dysregulated podocyte phenotype appears to be irreversible and largely unresponsive to standard-of-care FSGS therapies highlighting the urgent need for a nuanced understanding of disease evolution, novel therapeutics, and early intervention (4).
Normal Podocyte Function at the Glomerular Filtration Barrier
Podocytes are an essential cellular component of the tripartite glomerular filtration barrier (16). Podocytes are post-mitotic epithelial cells characterized by their highly specialized, actin-based membranous extentions known as foot processes (16). Podocytes envelop glomerular capillaries, adhering to the glomerular basement membrane (GBM) through a network of intermolecular interactions connecting foot processes to the GBM (16). Between interdigitating foot processes, a zipper-like assembly of proteins known as the slit-diaphragm functions as a molecular seive to provide charge and size selectivity for ultrafiltration (17–19). Podocytes produce the molecular constituents of the slit diaphragm and the expression of these proteins coincides with podocyte differentiation and maturation (20, 21). For example, the slit diaphragm protein nephrin, first identified in a Finnish cohort study of congenital nephrotic syndrome (17, 22), is produced by podocytes. The various roles of nephrin at the slit diaphram and as a modulator of prosurvival signaling in podocytes are well-documented (23–28) and will not be detailed in this review, however, it is clear that disease processes that impair podocyte nephrin expression, and other slit diaphragm components, result in podocyte dysfunction and drive the development of proteinuria and the FSGS lesion (29).
Effects of Viral Gene Expression on Podocyte Phyisology and Function
The collapsing FSGS lesion of HIVAN is characterized by podocyte dedifferentiation and hyperplasia, loss of podocyte maturity markers, foot process effacement and podocyte detachment, podocyte apoptosis, and heavy proteinuria (30–33). The cytotoxic effects of HIV gene expression in podocytes are well-established, and experimental models of HIVAN suggest that podocyte-restricted expression of viral proteins is sufficient to induce a dysegulated podocyte phenotype and the collapsing FSGS lesion (30, 31, 34–38). In particular, subtantial evidence exists for the roles of the HIV proteins Nef and Vpr in driving podocyte injury and dysfunction in HIVAN (Figures 1, 2).
Nef
Nef is one of four accesory proteins (i.e., Nef, Vpr, Vif, and Vpu) expressed by HIV (39). Despite early descriptions of Nef as a negative regulatory factor of viral replication (40–42), subsequent studies demonstrated that Nef exerts a neutral or positive effect on viral replication in various cell types (39, 42). Although HIV does not appear to produce productive infection in podocytes (13, 43), Nef exerts a variety of deleterious effects on podocyte physiology and function that are unrelated to the enhancement of viral replication. In 2002, Husain et al. demonstrated that Nef expression induced the loss of maturity markers, proliferation and anchorage-independent growth in cultured human podocytes (36). These data were later validated in a murine model of podocyte-restricted Nef expression. Husain et al. showed that podocyte-specific expression of Nef caused the loss of maturity marker expression (i.e., synaptopodin and WT1), induction of STAT3 activtion, and expression of the proliferation marker Ki-67 (36). Notably, this model did not manifest the proteinuria or glomerular injury chracteristic of HIVAN, leading the authors to conclude that Nef may be responsible for the early molecular changes that drive podocyte injury in HIVAN. Sunamoto et al. demonstrated that Nef expression was necessary and sufficient to induce proliferation and dedifferentiation in murine podocytes (44). He et al. later provided mechanistic insights into the role of Nef in podocyte hyperplasia when they demonstrated that Nef stimulates pro-proliferative signaling through the Src tyrosine kinase-dependent activation of Ras-c-Raf-MAPK1/2 and STAT3 signaling in conditionally immortalized human poocytes (45). The importance of STAT3 activation in podocyte hyperplasia was highlighted by the work of Feng et al. who demonstrated that reduction of STAT3 expression and activity ameliorated proteinuria, glomerulosclerosis, and tubulointersitial injury in a murine model of HIVAN (46). Similar findings were also reported by Gu et al. with STAT3 gene deletion in the same animal model (47). STAT3 is an established transcriptional regulator of molecules that drive cell-cycle re-entry and proliferation such as C-Myc, Cyclin D-1, CDC25A, and anillin (48, 49), supporting the hypothesis that STAT3 is a key regulator of podocyte proliferation in HIVAN. Several studies have also implicated Nef in the disruption of the podocyte cytoskeleton through various intermolecular interactions with actin and other key regulators of cytoskeletal dynamics (50–55). Other functions of Nef, such as its ability to interact with clathrin at the plasma membrane to disrupt endocytic trafficking, may also contribute to podocyte injury; however, this aspect of Nef signaling has not been documented in podocytes.
Vpr
The HIV accessory protein Viral Protein R (Vpr) has also been identified as a significant contributor to kidney injury in HIVAN. Like Nef, podocyte-restricted expression of Vpr in murine models established on the susceptible FVB/N background was sufficient to produce glomerular collapse and tubulointerstitial disease (38, 56, 57). Double transgenic expression of Vpr and Nef synergistically induced the full spectrum of podocyte injury, glomerular collapse, and tubulointerstitial diseased observed in human HIVAN (38, 57). In renal tubular epithelial cells (RTECs), Vpr has been shown to induce G2/M phase cell cycle arrest and dysregulation of cytokinesis (57–60). Vpr also induces apotosis in RTECs via the persistant activation of ERK MAP kinase and the upregulation of the ubiquitin-like protein FAT10 (61, 62). Less is known about the mechanisms of Vpr-induced podocyte injury. In 2014, Gbadegesin et al. demonstrated that the cytokinesis regulatory protein and pro-proliferative signaling molecule anillin, was upregulated in a murine model of podocyte-restricted Vpr expression (63). Anillin is an essential component of the cytokinetic ring and a driver of abnormal cellular proliferation in various malignacies (64–66). In the Vpr transgenic mouse, the upregulation of anillin in glomerular podocytes likely represents an accumulation of anillin in arrested cells or a cell-type specific derangement of cytokinetic drive and cell-cycle re-entry signaling.
Other Viral Proteins
The HIV regulatory protein Tat may also contribute to podocyte dysfunction in HIVAN. Tat is essential for HIV gene transactivation (67). In primary and conditionally immortalized podocytes, Conaldi et al. showed that Tat expression induced basic fibroblast growth factor (FGF-2)-driven hyperplasia, loss of maturity markers, cytoskeletal dysregulation, and impairment of permselectivity in a dose-dependent manner (43). Similar findings were later reported by Doublier et al. who showed that Tat exposure impaired the permeability of isolated glomeruli and reduced nephrin expression in conditionally immortalized human podocytes (68). Insights into the mechanisms of Tat-induced podocyte injury were provided by Xie et al., who reported that Tat targets to cholesterol-enriched lipid rafts, where it drives RhoA, matrix metalloproteinase-9 expression and FGF-2-mediated proproliferative signaling (69). Notably, murine models of podocyte-restricted Tat expression have failed to recapitulate the HIVAN phenotype (70). Overexpression of other HIV proteins such as Rev, Vif, and Vpu have not been associated with podocyte cytotoxicity and have not induced glomerular injury in murine models (70).
Contributions of the High-Risk Apolipoprotein L1 (APOL1) Alleles
The epidemiology of HIVAN is also consistent with a role for host genetic susceptibility, with a marked predilection for individuals of African descent. The discovery of high-risk variants in the APOL1 gene provided evidence of a genetic contribution to the racial disparity (71). The G1 (rs4821481 and rs3752462) and G2 (rs71785313) APOL1 variants were identified in an association analysis comparing 205 African-American individuals with non-familial, biopsy-proven FSGS and 180 healthy African-American controls. APOL1 encodes apolipoprotein L1, a trypanolytic serum factor that confers resistance against the parasitic infection that causes African sleeping sickness (71). The G1 and G2 APOL1 variants are found exclusively in individuals of recent African descent and confer resistance against a deadly subspecies of Trypanosoma that is normally resistant to lysis by wild-type APOL1. Carrying two APOL1 variants significantly enhances the risk of developing HIVAN in untreated HIV-infected individuals and explains up to 35% of the disease (72, 73). Our understanding of the mechanisms of APOL1-mediated kidney injury is rapidly increasing. In 2016, Olabisi et al. identified direct cytotoxic effects of the APOL1 proteins via the formation of cation permeable pores that disrupt potassium flux and lead to cellular swelling and death (74). Subsequently, Jha et al showed that the APOL1-mediated enhancement of potassium efflux induces proinflammatory cytokine expression, activation of the NLRP3 inflammasome and cellular pyroptosis (75). Other mechanisms of APOL1-mediated cellular injury have been uncovered. For example, Ma et al demonstrated that the APOL1 renal risk variants induce mitochondrial fission, reduce mitochondrial repiratory capacity, respiration rate and membrane potential (76, 77). Expression of the G1 and G2 variants also induced dysregulation of endosomal trafficking and lysosomal acidification in Drosophila and Saccharomyces (78). Additionally, the G1 and G2 renal risk variants have been shown to enhance the expression of miR193a, a negative regulator of autophagy (79). Consistent with an impairment in autophagy, Wen et al showed that overexpression of the APOL1 risk alleles induce endoplasmic reticulum stress in culture human podocytes (80). Upregulation of miR193a has also been shown to impair adherens complex stability, disrupt actomyosin cytoskeletal organization, reduce nephrin expression and promote dedifferentiation in podocytes (79, 81).
In the context of HIVAN, elaboration of interferon-γ (INF-γ) and other circulating mediators of innate immunity signaling appear to drive APOL1 gene transcription (82, 83). In vitro, the cytotoxicity of the high-risk APOL1 variants is dose-dependent, suggesting that any process that enhances the expression of the APOL1 renal risk alleles may provoke glomerular injury (84). This finding may, at least partially, explain why HIV is among the strongest promoters of glomerular disease in the setting of the high-risk APOL1 genotype, which has been associated with up to 89-fold increase in odds of HIVAN (85). Notably, collapsing glomerulopathy has been observed in individuals of African descent treated with interferon therapies and following viral infection with Parvovirus B19, CMV, EBV, HTLV1, Coxsackie B, Dengue, Zika, and most recently, SARS-CoV-2 (86–88). Small studies have demonstrated an association between the high-risk APOL1 genotype and the development of collapsing FSGS in the setting of COVID-19 (89) (Figures 1, 2).
Mechanisms of Proteinuria in Other Kidney Diseases in PLWH
HIV infection, by a variety of intracellular and systemic influences on podocyte physiology, perturbs cellular fate commitment, gene expression, and viability to promote development of the collapsing FSGS lesion of HIVAN. A nuanced understanding of the processes that drive podocyte injury in HIVAN may uncover novel therapeutic targets for treatment of other glomerular diseases. Prompted by epidemiologic studies demonstrating accelerated progression of kidney disease in the setting of HIV and diabetes (90), Mallipattu et al. demonstrated that the induction of diabetes with streptozotocin resulted in more prominent histologic changes in HIV-transgenic mice compared to wild-type littermates (91). These findings were confirmed in a subsequent study using podocyte-specific transgenic mice with low HIV transgene expression to more closely reflect the current clinical status of ART-treated individuals (92). In this model, HIV and diabetes had a synergistic effect on the expression of Sirtuin-1 deacetylase, suggesting a potential therapeutic role for Sirtuin-1 agonists.
A lack of animal models has slowed progress toward elucidating the pathogenesis of immune complex glomerular disease in PLWH. This has been compounded by the diverse spectrum of glomerular lesions that occur in this setting and that have been considered together in most clinical studies. Small but rigorous human studies have suggested a role for immune complexes directed against HIV antigens in the pathogenesis of immune complex kidney disease (93). Because podocytes have been shown to play a role in the clearance of immune deposits (94), it is possible that HIV-induced podoycte damage also promotes immune complex kidney disease in PLWH.
Kidney injury due to the antiretroviral agent tenofovir disoproxil fumarate (TDF) may also present with proteinuria, although this is typically low molecular weight proteinuria rather than albuminuria. Tenofovir is a nucleotide analog that is chemically related to the older antiviral agents cidofovir and adefovir, both of which are known to exhibit dose-limiting proximal tubular toxicity. The first approved tenofovir prodrug, TDF, has been associated with proximal tubulopathy and non-albumin proteinuria. Although the mechanism of proximal tubular cell injury has not been fully elucidated, it is thought to involve mitochondrial dysfunction as a result of the weak inhibition of mitochondrial DNA polymerase gamma (95, 96). Tenofovir is eliminated by glomerular filtration and active proximal tubular cell secretion, and an increase in intracellular concentration due to increased plasma concentration, decreased glomerular filtration, or impaired apical transport of tenofovir is thought to increase the risk of proximal tubular cell dysfunction or injury. Although it is possible that HIV-induced cell damage promotes tenofovir toxicity, non-albumin proteinuria has also been observed with the use of TDF for HIV pre-exposure prophylaxis in HIV-negative individuals (97). A newer prodrug, tenofovir alafenamide, is effective at lower plasma concentrations and may reduce the risk of tenofovir toxicity, although longer followup is needed.
Despite some risk of nephrotoxicity with tenofovir and other antiretroviral agents, the use of ART for treatment and prevention of HIV infection is currently the most effective way to mitigate the myriad pathogenic effects of HIV on the kidneys. While the incidence of advanced kidney disease due to HIVAN has decreased with the use of ART, HIVAN remains a valuable model for the study of podocyte injury and APOL1-induced glomerular disease.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
GH was supported by the NIH/NIDDK (K08DK111940), the American Society of Nephrology and the Harold Amos Medical Faculty Development Program, the Northwestern University George M. O'Brien Kidney Research Award, the Doris Duke Charitable Foundation, and the Duke Claude D. Pepper Older Americans Independence Center. CW was supported by the NIH/NIDDK (R01DK112258 and P01DK056492).
Conflict of Interest
GH is also a consultant for Reata Pharmaceuticals, Travere Pharmaceuticals, Otsuka Pharmaceuticals and Goldfinch Bio.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. UNAIDS. UNAIDS Global HIV & AIDS Statistics (2021). Available online at: https://www.unaids.org/en/resources/fact-sheet (accessed July 14, 2021).
2. Pardo V, Aldana M, Colton RM, Fischl MA, Jaffe D, Moskowitz L, et al. Glomerular lesions in the acquired immunodeficiency syndrome. Ann Intern Med. (1984) 101:429–34. doi: 10.7326/0003-4819-101-4-429
3. Rao TK, Filippone EJ, Nicastri AD, Landesman SH, Frank E, Chen CK, et al. Associated focal and segmental glomerulosclerosis in the acquired immunodeficiency syndrome. N Engl J Med. (1984) 310:669–73. doi: 10.1056/NEJM198403153101101
4. Kudose S, Santoriello D, Bomback AS, Stokes MB, Batal I, Markowitz GS, et al. The spectrum of kidney biopsy findings in HIV-infected patients in the modern era. Kidney Int. (2020) 97:1006–16. doi: 10.1016/j.kint.2020.01.018
5. Cohen AH, Sun NC, Shapshak P, Imagawa DT. Demonstration of human immunodeficiency virus in renal epithelium in HIV-associated nephropathy. Mod Pathol. (1989) 2:125–8.
6. Kimmel PL, Ferreira-Centeno A, Farkas-Szallasi T, Abraham AA, Garrett CT. Viral DNA in microdissected renal biopsy tissue from HIV infected patients with nephrotic syndrome. Kidney Int. (1993) 43:1347–52. doi: 10.1038/ki.1993.189
7. Tanji N, Ross MD, Tanji K, Bruggeman LA, Markowitz GS, Klotman PE, et al. Detection and localization of HIV-1 DNA in renal tissues by in situ polymerase chain reaction. Histol Histopathol. (2006) 21:393–401. doi: 10.14670/hh-21.393
8. Marras D, Bruggeman LA, Gao F, Tanji N, Mansukhani MM, Cara A, et al. Replication and compartmentalization of HIV-1 in kidney epithelium of patients with HIV-associated nephropathy. Nat Med. (2002) 8:522–6. doi: 10.1038/nm0502-522
9. Eitner F, Cui Y, Hudkins KL, Stokes MB, Segerer S, Mack M, et al. Chemokine receptor CCR5 and CXCR4 expression in HIV-associated kidney disease. J Am Soc Nephrol. (2000) 11:856–67. doi: 10.1681/ASN.V115856
10. Ray N, Doms RW. HIV-1 coreceptors and their inhibitors. Curr Top Microbiol Immunol. (2006) 303:97–120. doi: 10.1007/978-3-540-33397-5_5
11. Alkhatib G. The biology of CCR5 and CXCR4. Curr Opin HIV AIDS. (2009) 4:96–103. doi: 10.1097/COH.0b013e328324bbec
12. Mikulak J, Singhal PC. HIV-1 entry into human podocytes is mediated through lipid rafts. Kidney Int. (2010) 77:72–3; author reply 73–74. doi: 10.1038/ki.2009.366
13. Khatua AK, Taylor HE, Hildreth JE, Popik W. Non-productive HIV-1 infection of human glomerular and urinary podocytes. Virology. (2010) 408:119–27. doi: 10.1016/j.virol.2010.09.005
14. Li J, Das JR, Tang P, Han Z, Jaiswal JK, Ray PE. Transmembrane TNF-alpha facilitates HIV-1 infection of podocytes cultured from children with HIV-associated nephropathy. J Am Soc Nephrol. (2017) 28:862–75. doi: 10.1681/ASN.2016050564
15. Daecke J, Fackler OT, Dittmar MT, Krausslich HG. Involvement of clathrin-mediated endocytosis in human immunodeficiency virus type 1 entry. J Virol. (2005) 79:1581–94. doi: 10.1128/JVI.79.3.1581-1594.2005
16. Arif E, Nihalani D. Glomerular filtration barrier assembly: an insight. Postdoc J. (2013) 1:33–45. doi: 10.14304/SURYA.JPR.V1N4.4
17. Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, et al. Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol Cell. (1998) 1:575–82. doi: 10.1016/S1097-2765(00)80057-X
18. Wartiovaara J, Ofverstedt LG, Khoshnoodi J, Zhang J, Makela E, Sandin S, et al. Nephrin strands contribute to a porous slit diaphragm scaffold as revealed by electron tomography. J Clin Invest. (2004) 114:1475–83. doi: 10.1172/JCI22562
19. Gagliardini E, Conti S, Benigni A, Remuzzi G, Remuzzi A. Imaging of the porous ultrastructure of the glomerular epithelial filtration slit. J Am Soc Nephrol. (2010) 21:2081–9. doi: 10.1681/ASN.2010020199
20. Holthofer H. Molecular architecture of the glomerular slit diaphragm: lessons learnt for a better understanding of disease pathogenesis. Nephrol Dial Transplant. (2007) 22:2124–8. doi: 10.1093/ndt/gfm344
21. Kreidberg JA. Podocyte differentiation and glomerulogenesis. J Am Soc Nephrol. (2003) 14:806–14. doi: 10.1097/01.ASN.0000054887.42550.14
22. Tryggvason K, Ruotsalainen V, Wartiovaara J. Discovery of the congenital nephrotic syndrome gene discloses the structure of the mysterious molecular sieve of the kidney. Int J Dev Biol. (1999) 43:445–51.
23. Li X, Chuang PY, D'Agati VD, Dai Y, Yacoub R, Fu J, et al. Nephrin preserves podocyte viability and glomerular structure and function in adult kidneys. J Am Soc Nephrol. (2015) 26:2361–77. doi: 10.1681/ASN.2014040405
24. Patrakka J, Tryggvason K. Nephrin–a unique structural and signaling protein of the kidney filter. Trends Mol Med. (2007) 13:396–403. doi: 10.1016/j.molmed.2007.06.006
25. Gigante M, Piemontese M, Gesualdo L, Iolascon A, Aucella F. Molecular and genetic basis of inherited nephrotic syndrome. Int J Nephrol. (2011) 2011:792195. doi: 10.4061/2011/792195
26. New LA, Martin CE, Scott RP, Platt MJ, Keyvani Chahi A, Stringer CD, et al. Nephrin tyrosine phosphorylation is required to stabilize and restore podocyte foot process architecture. J Am Soc Nephrol. (2016) 27:2422–35. doi: 10.1681/ASN.2015091048
27. Martin CE, Jones N. Nephrin signaling in the podocyte: an updated view of signal regulation at the slit diaphragm and beyond. Front Endocrinol (Lausanne). (2018) 9:302. doi: 10.3389/fendo.2018.00302
28. Verma R, Venkatareddy M, Kalinowski A, Li T, Kukla J, Mollin A, et al. Nephrin is necessary for podocyte recovery following injury in an adult mature glomerulus. PLoS ONE. (2018) 13:e0198013. doi: 10.1371/journal.pone.0198013
29. Testagrossa L, Azevedo Neto R, Resende A, Woronik V, Malheiros D. Immunohistochemical expression of podocyte markers in the variants of focal segmental glomerulosclerosis. Nephrol Dial Transplant. (2013) 28:91–8. doi: 10.1093/ndt/gfs325
30. Barisoni L, Kriz W, Mundel P, D'Agati V. The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. (1999) 10:51–61. doi: 10.1681/ASN.V10151
31. Yang Y, Gubler MC, Beaufils H. Dysregulation of podocyte phenotype in idiopathic collapsing glomerulopathy and HIV-associated nephropathy. Nephron. (2002) 91:416–23. doi: 10.1159/000064281
32. Barisoni L, Mokrzycki M, Sablay L, Nagata M, Yamase H, Mundel P. Podocyte cell cycle regulation and proliferation in collapsing glomerulopathies. Kidney Int. (2000) 58:137–43. doi: 10.1046/j.1523-1755.2000.00149.x
33. Canaud G, Dejucq-Rainsford N, Avettand-Fenoel V, Viard JP, Anglicheau D, Bienaime F, et al. The kidney as a reservoir for HIV-1 after renal transplantation. J Am Soc Nephrol. (2014) 25:407–19. doi: 10.1681/ASN.2013050564
34. Barisoni L, Bruggeman LA, Mundel P, D'Agati VD, Klotman PE. HIV-1 induces renal epithelial dedifferentiation in a transgenic model of HIV-associated nephropathy. Kidney Int. (2000) 58:173–81. doi: 10.1046/j.1523-1755.2000.00152.x
35. Bruggeman LA, Dikman S, Meng C, Quaggin SE, Coffman TM, Klotman PE. Nephropathy in human immunodeficiency virus-1 transgenic mice is due to renal transgene expression. J Clin Invest. (1997) 100:84–92. doi: 10.1172/JCI119525
36. Husain M, D'Agati VD, He JC, Klotman ME, Klotman PE. HIV-1 Nef induces dedifferentiation of podocytes in vivo: a characteristic feature of HIVAN. AIDS. (2005) 19:1975–80. doi: 10.1097/01.aids.0000191918.42110.27
37. Zhong J, Zuo Y, Ma J, Fogo AB, Jolicoeur P, Ichikawa I, et al. Expression of HIV-1 genes in podocytes alone can lead to the full spectrum of HIV-1-associated nephropathy. Kidney Int. (2005) 68:1048–60. doi: 10.1111/j.1523-1755.2005.00497.x
38. Zuo Y, Matsusaka T, Zhong J, Ma J, Ma LJ, Hanna Z, et al. HIV-1 genes vpr and nef synergistically damage podocytes, leading to glomerulosclerosis. J Am Soc Nephrol. (2006) 17:2832–43. doi: 10.1681/ASN.2005080878
40. Terwilliger E, Sodroski JG, Rosen CA, Haseltine WA. Effects of mutations within the 3' orf open reading frame region of human T-cell lymphotropic virus type III (HTLV-III/LAV) on replication and cytopathogenicity. J Virol. (1986) 60:754–60. doi: 10.1128/jvi.60.2.754-760.1986
41. Ahmad N, Venkatesan S. Nef protein of HIV-1 is a transcriptional repressor of HIV-1 LTR. Science. (1988) 241:1481–5. doi: 10.1126/science.3262235
42. Cheng-Mayer C, Iannello P, Shaw K, Luciw PA, Levy JA. Differential effects of nef on HIV replication: implications for viral pathogenesis in the host. Science. (1989) 246:1629–32. doi: 10.1126/science.2531920
43. Conaldi PG, Bottelli A, Baj A, Serra C, Fiore L, Federico G, et al. Human immunodeficiency virus-1 tat induces hyperproliferation and dysregulation of renal glomerular epithelial cells. Am J Pathol. (2002) 161:53–61. doi: 10.1016/S0002-9440(10)64156-9
44. Sunamoto M, Husain M, He JC, Schwartz EJ, Klotman PE. Critical role for Nef in HIV-1-induced podocyte dedifferentiation. Kidney Int. (2003) 64:1695–701. doi: 10.1046/j.1523-1755.2003.00283.x
45. He JC, Husain M, Sunamoto M, D'Agati VD, Klotman ME, Iyengar R, et al. Nef stimulates proliferation of glomerular podocytes through activation of Src-dependent Stat3 and MAPK1,2 pathways. J Clin Invest. (2004) 114:643–51. doi: 10.1172/JCI200421004
46. Feng X, Lu TC, Chuang PY, Fang W, Ratnam K, Xiong H, et al. Reduction of Stat3 activity attenuates HIV-induced kidney injury. J Am Soc Nephrol. (2009) 20:2138–46. doi: 10.1681/ASN.2008080879
47. Gu L, Dai Y, Xu J, Mallipattu S, Kaufman L, Klotman PE, et al. Deletion of podocyte STAT3 mitigates the entire spectrum of HIV-1-associated nephropathy. AIDS. (2013) 27:1091–8. doi: 10.1097/QAD.0b013e32835f1ea1
48. Carpenter RL, Lo HW. STAT3 target genes relevant to human cancers. Cancers (Basel). (2014) 6:897–925. doi: 10.3390/cancers6020897
49. Hall G, Lane BM, Khan K, Pediaditakis I, Xiao J, Wu G, et al. The human FSGS-causing ANLN R431C mutation induces dysregulated PI3K/AKT/mTOR/Rac1 signaling in podocytes. J Am Soc Nephrol. (2018) 29:2110–22. doi: 10.1681/ASN.2017121338
50. Lu TC, He JC, Wang ZH, Feng X, Fukumi-Tominaga T, Chen N, et al. HIV-1 Nef disrupts the podocyte actin cytoskeleton by interacting with diaphanous interacting protein. J Biol Chem. (2008) 283:8173–82. doi: 10.1074/jbc.M708920200
51. Tan R, Patni H, Tandon P, Luan L, Sharma B, Salhan D, et al. Nef interaction with actin compromises human podocyte actin cytoskeletal integrity. Exp Mol Pathol. (2013) 94:51–7. doi: 10.1016/j.yexmp.2012.06.001
52. Lee CH, Saksela K, Mirza UA, Chait BT, Kuriyan J. Crystal structure of the conserved core of HIV-1 Nef complexed with a Src family SH3 domain. Cell. (1996) 85:931–42. doi: 10.1016/S0092-8674(00)81276-3
53. Geyer M, Fackler OT, Peterlin BM. Structure–function relationships in HIV-1 Nef. EMBO Rep. (2001) 2:580–5. doi: 10.1093/embo-reports/kve141
54. Arold ST, Baur AS. Dynamic Nef and Nef dynamics: how structure could explain the complex activities of this small HIV protein. Trends Biochem Sci. (2001) 26:356–63. doi: 10.1016/S0968-0004(01)01846-1
55. Ratnam KK, He CJ, Klotman P. Nef as a Proliferative Factor for Kidney Epithelial Cells in HIV-Associated Nephropathy. Clin Med Oncol. (2008) 2:539–45. doi: 10.4137/CMO.S661
56. Hiramatsu N, Hiromura K, Shigehara T, Kuroiwa T, Ideura H, Sakurai N, et al. Angiotensin II type 1 receptor blockade inhibits the development and progression of HIV-associated nephropathy in a mouse model. J Am Soc Nephrol. (2007) 18:515–27. doi: 10.1681/ASN.2006030217
57. Rednor SJ, Ross MJ. Molecular Mechanisms of Injury in HIV-Associated Nephropathy. Front Med (Lausanne). (2018) 5:177. doi: 10.3389/fmed.2018.00177
58. Snyder A, Alsauskas ZC, Leventhal JS, Rosenstiel PE, Gong P, Chan JJ, et al. HIV-1 viral protein r induces ERK and caspase-8-dependent apoptosis in renal tubular epithelial cells. AIDS. (2010) 24:1107–19. doi: 10.1097/QAD.0b013e328337b0ab
59. Rosenstiel PE, Gruosso T, Letourneau AM, Chan JJ, LeBlanc A, Husain M, et al. HIV-1 Vpr inhibits cytokinesis in human proximal tubule cells. Kidney Int. (2008) 74:1049–58. doi: 10.1038/ki.2008.303
60. Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. (2010) 16:535–43. doi: 10.1038/nm.2144
61. Snyder A, Alsauskas Z, Gong P, Rosenstiel PE, Klotman ME, Klotman PE, et al. FAT10: a novel mediator of Vpr-induced apoptosis in human immunodeficiency virus-associated nephropathy. J Virol. (2009) 83:11983–8. doi: 10.1128/JVI.00034-09
62. Ross MJ, Wosnitzer MS, Ross MD, Granelli B, Gusella GL, Husain M, et al. Role of ubiquitin-like protein FAT10 in epithelial apoptosis in renal disease. J Am Soc Nephrol. (2006) 17:996–1004. doi: 10.1681/ASN.2005070692
63. Gbadegesin RA, Hall G, Adeyemo A, Hanke N, Tossidou I, Burchette J, et al. Mutations in the gene that encodes the F-actin binding protein anillin cause FSGS. J Am Soc Nephrol. (2014) 25:1991–2002. doi: 10.1681/ASN.2013090976
64. Magnusson K, Gremel G, Ryden L, Ponten V, Uhlen M, Dimberg A, et al. ANLN is a prognostic biomarker independent of Ki-67 and essential for cell cycle progression in primary breast cancer. BMC Cancer. (2016) 16:904. doi: 10.1186/s12885-016-2923-8
65. Lian YF, Huang YL, Wang JL, Deng MH, Xia TL, Zeng MS, et al. Anillin is required for tumor growth and regulated by miR-15a/miR-16-1 in HBV-related hepatocellular carcinoma. Aging (Albany NY). (2018) 10:1884–901. doi: 10.18632/aging.101510
66. Tuan NM, Lee CH. Role of anillin in tumour: from a prognostic biomarker to a novel target. Cancers (Basel). (2020) 12:1600. doi: 10.3390/cancers12061600
67. Gatignol A, Duarte M, Daviet L, Chang YN, Jeang KT. Sequential steps in Tat trans-activation of HIV-1 mediated through cellular DNA, RNA, and protein binding factors. Gene Expr. (1996) 5:217–28.
68. Doublier S, Zennaro C, Spatola T, Lupia E, Bottelli A, Deregibus MC, et al. HIV-1 Tat reduces nephrin in human podocytes: a potential mechanism for enhanced glomerular permeability in HIV-associated nephropathy. AIDS. (2007) 21:423–32. doi: 10.1097/QAD.0b013e328012c522
69. Xie X, Colberg-Poley AM, Das JR, Li J, Zhang A, Tang P, et al. The basic domain of HIV-tat transactivating protein is essential for its targeting to lipid rafts and regulating fibroblast growth factor-2 signaling in podocytes isolated from children with HIV-1-associated nephropathy. J Am Soc Nephrol. (2014) 25:1800–13. doi: 10.1681/ASN.2013070710
70. Rosenstiel P, Gharavi A, D'Agati V, Klotman P. Transgenic and infectious animal models of HIV-associated nephropathy. J Am Soc Nephrol. (2009) 20:2296–304. doi: 10.1681/ASN.2008121230
71. Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. (2010) 329:841–5. doi: 10.1126/science.1193032
72. Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. (2011) 22:2129–37. doi: 10.1681/ASN.2011040388
73. Friedman DJ, Pollak MR. Apolipoprotein L1 and kidney disease in African Americans. Trends Endocrinol Metab. (2016) 27:204–15. doi: 10.1016/j.tem.2016.02.002
74. Olabisi OA, Zhang JY, VerPlank L, Zahler N, DiBartolo S, Heneghan JF, et al. APOL1 kidney disease risk variants cause cytotoxicity by depleting cellular potassium and inducing stress-activated protein kinases. Proc Natl Acad Sci USA. (2016) 113:830–7. doi: 10.1073/pnas.1522913113
75. Bruno J, Pozzi N, Oliva J, Edwards JC. Apolipoprotein L1 confers pH-switchable ion permeability to phospholipid vesicles. J Biol Chem. (2017) 292:18344–53. doi: 10.1074/jbc.M117.813444
76. Schaub C, Verdi J, Lee P, Terra N, Limon G, Raper J, et al. Cation channel conductance and pH gating of the innate immunity factor APOL1 are governed by pore-lining residues within the C-terminal domain. J Biol Chem. (2020) 295:13138–49. doi: 10.1074/jbc.RA120.014201
77. Giovinazzo JA, Thomson RP, Khalizova N, Zager PJ, Malani N, Rodriguez-Boulan E, et al. Apolipoprotein L-1 renal risk variants form active channels at the plasma membrane driving cytotoxicity. Elife. (2020) 9:e51185. doi: 10.7554/eLife.51185
78. Lan X, Jhaveri A, Cheng K, Wen H, Saleem MA, Mathieson PW, et al. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am J Physiol Renal Physiol. (2014) 307:F326–36. doi: 10.1152/ajprenal.00647.2013
79. Kruzel-Davila E, Shemer R, Ofir A, Bavli-Kertselli I, Darlyuk-Saadon I, Oren-Giladi P, et al. APOL1-mediated cell injury involves disruption of conserved trafficking processes. J Am Soc Nephrol. (2017) 28:1117–30. doi: 10.1681/ASN.2016050546
80. Wen H, Kumar V, Lan X, Shoshtari SSM, Eng JM, Zhou X, et al. APOL1 risk variants cause podocytes injury through enhancing endoplasmic reticulum stress. Biosci Rep. (2018) 38:BSR20171713. doi: 10.1042/BSR20171713
81. Wakashin H, Heymann J, Roshanravan H, Daneshpajouhnejad P, Rosenberg A, Shin MK, et al. APOL1 renal risk variants exacerbate podocyte injury by increasing inflammatory stress. BMC Nephrol. (2020) 21:371. doi: 10.1186/s12882-020-01995-3
82. Nichols B, Jog P, Lee JH, Blackler D, Wilmot M, D'Agati V, et al. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int. (2015) 87:332–42. doi: 10.1038/ki.2014.270
83. Kopp JB, Heymann J, Winkler CA. APOL1 renal risk variants: fertile soil for HIV-associated nephropathy. Semin Nephrol. (2017) 37:514–9. doi: 10.1016/j.semnephrol.2017.07.004
84. Datta S, Kataria R, Zhang JY, Moore S, Petitpas K, Mohamed A, et al. Kidney disease-associated APOL1 variants have dose-dependent, dominant toxic gain-of-function. J Am Soc Nephrol. (2020) 31:2083–96. doi: 10.1681/ASN.2020010079
85. Kasembeli AN, Duarte R, Ramsay M, Mosiane P, Dickens C, Dix-Peek T, et al. APOL1 risk variants are strongly associated with HIV-associated nephropathy in Black South Africans. J Am Soc Nephrol. (2015) 26:2882–90. doi: 10.1681/ASN.2014050469
86. Markowitz GS, Nasr SH, Stokes MB, D'Agati VD. Treatment with IFN-{alpha}, -{beta}, or -{gamma} is associated with collapsing focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. (2010) 5:607–15. doi: 10.2215/CJN.07311009
87. Chandra P, Kopp JB. Viruses and collapsing glomerulopathy: a brief critical review. Clin Kidney J. (2013) 6:1–5. doi: 10.1093/ckj/sft002
88. Nasr SH, Kopp JB. COVID-19-associated collapsing glomerulopathy: an emerging entity. Kidney Int Rep. (2020) 5:759–61. doi: 10.1016/j.ekir.2020.04.030
89. Wu H, Larsen CP, Hernandez-Arroyo CF, Mohamed MMB, Caza T, Sharshir M, et al. AKI and collapsing glomerulopathy associated with COVID-19 and APOL 1 high-risk genotype. J Am Soc Nephrol. (2020) 31:1688–95. doi: 10.1681/ASN.2020050558
90. Medapalli RK, Parikh CR, Gordon K, Brown ST, Butt AA, Gibert CL, et al. Comorbid diabetes and the risk of progressive chronic kidney disease in HIV-infected adults: data from the Veterans Aging Cohort Study. J Acquir Immune Defic Syndr. (2012) 60:393–9. doi: 10.1097/QAI.0b013e31825b70d9
91. Mallipattu SK, Liu R, Zhong Y, Chen EY, D'Agati V, Kaufman L, et al. Expression of HIV transgene aggravates kidney injury in diabetic mice. Kidney Int. (2013) 83:626–34. doi: 10.1038/ki.2012.445
92. Feng J, Bao L, Wang X, Li H, Chen Y, Xiao W, et al. Low expression of HIV genes in podocytes accelerates the progression of diabetic kidney disease in mice. Kidney Int. (2021) 99:914–25. doi: 10.1016/j.kint.2020.12.012
93. Kimmel PL, Phillips TM, Ferreira-Centeno A, Farkas-Szallasi T, Abraham AA, Garrett CT. HIV-associated immune-mediated renal disease. Kidney Int. (1993) 44:1327–40. doi: 10.1038/ki.1993.386
94. Alexander JJ, Wang Y, Chang A, Jacob A, Minto AW, Karmegam M, et al. Mouse podocyte complement factor H: the functional analog to human complement receptor 1. J Am Soc Nephrol. (2007) 18:1157–66. doi: 10.1681/ASN.2006101125
95. Herlitz LC, Mohan S, Stokes MB, Radhakrishnan J, D'Agati VD, Markowitz GS. Tenofovir nephrotoxicity: acute tubular necrosis with distinctive clinical, pathological, mitochondrial abnormalities. Kidney Int. (2010) 78:1171–7. doi: 10.1038/ki.2010.318
96. Lebrecht D, Venhoff AC, Kirschner J, Wiech T, Venhoff N, Walker UA. Mitochondrial tubulopathy in tenofovir disoproxil fumarate-treated rats. J Acquir Immune Defic Syndr. (2009) 51:258–63. doi: 10.1097/QAI.0b013e3181a666eb
Keywords: podocyte, glomerular disease, HIVAN-associated nephropathy, APOL1, collapsing FSGS
Citation: Hall G and Wyatt CM (2021) Mechanisms of Proteinuria in HIV. Front. Med. 8:749061. doi: 10.3389/fmed.2021.749061
Received: 28 July 2021; Accepted: 09 September 2021;
Published: 13 October 2021.
Edited by:
Ilse Sofia Daehn, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Pravin C. Singhal, North Shore Long Island Jewish Health System, United StatesSaraladevi Naicker, University of the Witwatersrand, South Africa
Copyright © 2021 Hall and Wyatt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christina M. Wyatt, christina.wyatt@duke.edu